
Data
Gene data is in genes-in-rows format, comma-separated values.Take final_project_data.zip file from Data directory, and unzip to extract 3 files:
- pp5i_train.gr.csv (training data, 1.7 MB)
- pp5i_train_class.txt (training data classes)
- pp5i_test.gr.csv (test data, 0.6MB)
Instructions
Training data: file pp5i_train.gr.csv, with with 7070 genes (no Affy controls) for 69 samples. A separate file pp5i_train_class.txt has classes for each sample, in the order corresponding to the order of samples in pp5i_train.gr.csv. There are 5 classes, labelled EPD, JPA, MED, MGL, RHB.
Test data: file pp5i_test.gr.csv, with 23 unlabelled samples and same genes. You can assume that the class distribution is similar.
Your goal is to learn the best model from the training data and use it to predict the label (class) for each sample in test data. You will also need to write a paper describing your effort.
Randomization experiments showed that one can get about 10-12 (from 23) correct answers with random guessing.
The final grade will be a combination of effort (40%), presentation (30%), and the accuracy of prediction, measured as 3*(Number_correct_answers - 11). The maximum grade is 106.
Below are suggested steps for doing this experiment, but you can vary and improve on the suggested approach, as long as you produce a prediction for the test set and describe your results.
Important Hints
Be sure that you don't use the sample number as one of the predictors. Training data is ordered by class, so sample number will appear to be a good predictor on cross-validation, but it will not work on the test data!
One of the MED samples in the training data is very likely misclassified (by a human). So the best result you can expect to get on cross validation is one error (on a MED sample) out of 69. However, this should not affect your accuracy on the test set (all labels there are correct).
You can make all the runs from Weka GUI interface, but if you can learn a Unix shell, you can run these repeated experiments much easier from the shell. (Caution: weka cross-validation uses a random number seed which is different in GUI and in shell, so cross-validation results may be slightly different if you call Weka from shell than if you use Weka Explorer).
You can complete the project using only simple steps, but the more advanced steps will give you extra credit and probably higher accuracy.
The following steps suggest one way of finding the best model -- you are welcome to make improvements, where you think appropriate.
See also Questions and Answers at the end.
Step 1. Data Cleaning
Threshold both train and test data to a minimum value of 20, maximum of 16,000.Step 2. Selecting top genes by class
- remove from train data genes with fold differences across samples less than 2
- for each class, generate subsets with top 2,4,6,8,10,12,15,20,25, and 30 top genes with the highest T-value
Optional: for each class, select top genes using highest absolute T-value (i.e. also include genes with high negative T-value) - for each N=2,4,6,8,10,12,15,20,25,30 combine top genes for each class into one file (removing duplicates, if any) and call the resulting file pp5i_train.topN.gr.csv
- Add the class as the last column, remove sample no, transpose each file to "genes-in-columns" format and convert it to arff.
Step 3. Find the best classifier/best gene set combination
Use the following Weka classifiers:
- NaiveBayes
- J48
- IB1
- IBk (for each value of K=2, 3, 4)
- one more Weka classifier of your choice -- that can work with multiclass data.
a. For each classifier, using default settings, measure classifier accuracy on the training set using previously generated files with top N=2,4,6,8,10,12,15,20,25,30 genes.
For IBk, test accuracy with K=2, 3 and 4.
b. Select the model and the gene set with the lowest cross-validation error.
Optional: once you found the gene set with the lowest cross-validation error, you can vary 1-2 additional relevant parameters for each classifier to see if the accuracy will improve. E.g. for J4.8, you can vary reducedErrorPruning and binarySplits
c. Use the gene names from best train gene set and extract the data corresponding to these genes
from the test set.
d. Convert test set to genes-in-columns format.
e. Add a Class column with "?" values as the last column
Step 4. Generate predictions for the test set
You should now have the best train file, call it pp5i_train.bestN.csv, (with 69 samples and bestN number of genes for whatever bestN you found) and a corresponding test file, call it pp5i_test.bestN.csv, with the same genes and 23 test samples. The train file will have all Class values while the test file Class column will have only "?"
a. Convert test file to arff format (you should already have .arff for train file from Step 3).
Important: In Weka, the variable declarations should be exactly the same for test and train file.
To achieve that, change the Class entry in pp5i_test.bestN.arff header section to be the same
as in train file, i.e.
@attribute Class {MED,MGL,RHB,EPD,JPA}
b. Use the best train file and the matching test file and generate predictions for the test file class.
If you are using GUI, then
- select best train set under Preprocess tab
- click on Supplied test set option under Classify tab and specify the matching test set
- specify the appropriate classifier parameters, if any.
- click on Start to run the classifier. Because the classes are unknown ("?") in the test set, the confusion matrix will show all zeros.
- Right-click on the model name in the result list panel (see figure)
and select from submenu Visualize classifier errors
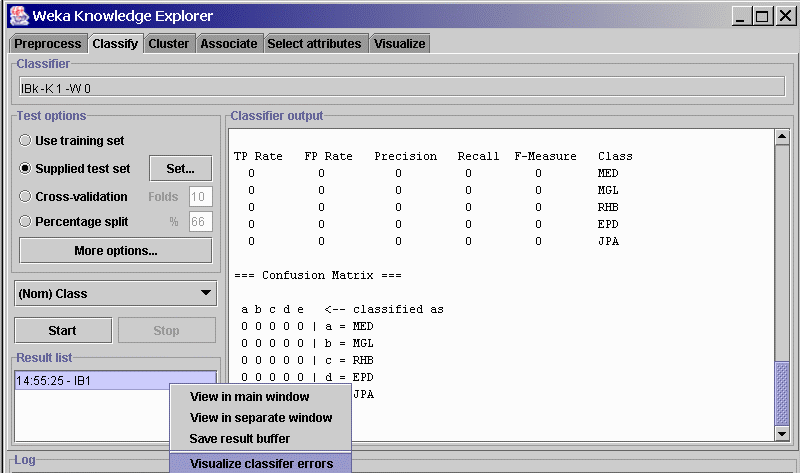
- From the visualization screen, select Save and Weka will save the test file and predictions in arff format.
- Extract from it a file with Instance_number and predictedClass columns and write them to a file *yourname*-predictions.csv
- You should have predictions for 23 instances, with instance number ranging from 0 to 22.
If you are using a shell, then you can generate predictions using, e.g.
java weka.classifiers.Classifier -t train_data.arff -T test.data.arff -p 0
Step 5. Write a paper describing your effort.
Document each step.
For each classifier used, give a paragraph describing this classifier.
Give a graph showing error rate versus number of genes.
Describe which classifier and which number of genes you have selected
Comment on the relative strengths and weaknesses of the classifiers you used for this type of data.
Questions and Answers
- Q: We understand how to compute T-value With two classes (e.g. ALL and AML). Avg1 would represent one and Avg2 would represent the other. How to do this with 5 classes?
A: For each class C, Avg1 (StDev1) would be the average (st. dev) gene expression value for samples in this class and Avg2 (Stdev2) -- for samples in other remaining classes.
- Q: How do we get a subset with top N genes with the highest T-value?
A: For each class, compute for each gene T-value as described above. Then take N genes with the highest T-value (you will have 5*N genes). Finally, combine the top genes for each class and remove duplicate genes.
- Q: when running Weka, we get "Train and test set are not compatible" error.
A: 1. Make sure that the train and test sets have EXACTLY the same attributes (fields),
including last entry:
@attribute Class {MED,MGL,RHB,EPD,JPA}
2. If the attribute descriptions appear to be identical but you still get the same error, you may have an invisible control character in one of the files. Remove the attribute descriptions from the test file and replace them with the attribute descriptions from the train file. That may help.
3. You can also run Weka from the command line, e.g.
java weka.classifiers.IB1 -t train_file.arff -T test_file.arff -p 0
- Q: Can we use experimenter environment?
A: Absolutely! It is specifically designed for the purpose of running multiple experiments. It is an advanced framework, and not required to do this project. However, if you are able to use it correctly, it will be favorably considered in grading.
- Q: Why do we get slightly different results in cross-validation when using experimenter environment and the GUI?
A: In Weka, each cross-validation run uses a random shuffling and I think that the experimenter environment uses a different random number seed than when you run from GUI one run at a time.
Slightly different results are normal in cross-validation, when it uses a random number to select the test set for each round.